Very low, low, and high-density lipoproteins (VLDL, LDL, HDL, respectively) are commonly measured on the standard blood chemistry panel as measures of cardiovascular disease risk. Not included on that panel is another lipoprotein, Lp(a), which is a modified form of LDL. What’s the relationship between Lp(a) with disease risk?
A meta-analysis of 36 studies that included 126,634 subjects reported that Lp(a) > 30 mg/dL (65 nmol/L) was significantly associated with an increased risk for heart attacks, coronary heart disease-related deaths, and ischemic strokes (Erqou et al. 2009):

Investigating further, of 2,100 candidate genes that were evaluated for predicting heart disease risk, genetic variation in the LPA gene was the strongest genetic risk factor (Clarke et al. 2009). Of the Lp(a)-related genes, SNPs for rs3798220 (increased risk allele = C) and rs10455872 (increased risk allele = G) were associated with a 92% and a 70% increased risk for coronary heart disease, respectively.
Based on these data, Lp(a) values less than 50 mg/dL (108 nmol/L) have been recommended, with 1-3 grams/day of niacin, which reduces Lp(a) levels, as the primary treatment for minimizing cardiovascular disease risk (Nordestgaard et al. 2010).
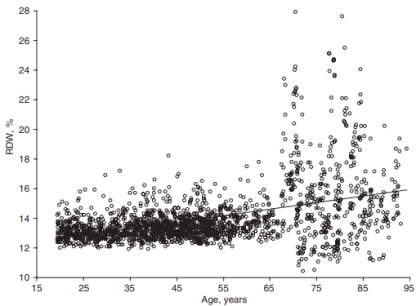
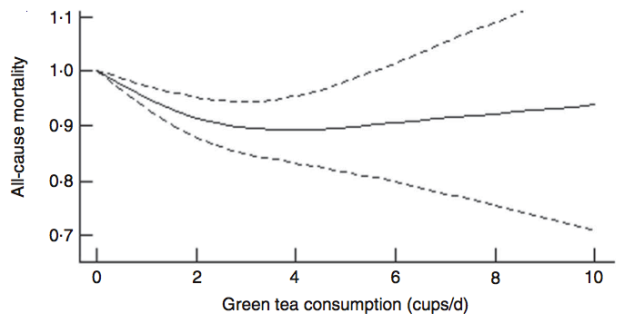
However, cardiovascular disease is only 1 outcome. What’s the data for Lp(a) and risk of death from all causes, not just cardiovascular disease-related deaths? In a study of 10,413 adults (average age, 55y), the lowest risk of death from all causes was reported for Lp(a) values of 270 mg/L (equivalent to 27 mg/dL, and 58 nmol/L). The log of 270 is 2.43, which corresponds to the lowest mortality risk on the chart below (Sawabe et al. 2012):

Interestingly, all-cause mortality risk was significantly increased only for Lp(a) values < 80 mg/L (log 80 = 1.90; equivalent to 17 nmol/L), when compared with intermediate (80 – 550 mg/L; log values from 1.9 – 2.7 on the chart; equivalent to 17 – 118 nmol/L) and high Lp(a) (> 550 mg/L; log values > 2.7 on the chart; equivalent to > 118 nmol/L).
In addition to low Lp(a) values, an increased risk of death from all causes (and a shorter lifespan) have also been reported for high Lp(a). When compared with Lp(a) < 21 nmol/L, Lp(a) > 199 nmol/L was associated with a 20% increased all-cause mortality risk (Langsted et al. 2019). In addition, median lifespan was 1.4 years shorter for subjects that had Lp(a) values > 199 nmol/L, when compared with < 21 nmol/L.
Based on the studies of Sawabe and Langsted, both low and high Lp(a) values may be bad for disease risk. What are my Lp(a) values?
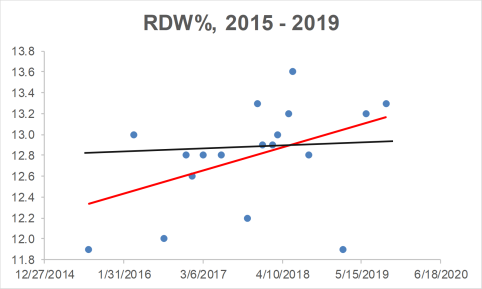
I’ve been tracking Lp(a) for the past 14 years, first, approximately 1x/year until I was 40, and second, 9 times since 2015, when I started daily nutrition tracking. In addition, I’ve measured it 4x in 2019, with the goal of getting it closer to the 58 nmol/L value of the Sawabe study. When I first started measuring Lp(a) in 2005, it was ~150 nmol/L, which is way higher than the < 65 nmol/L that was reported for reduced cardiovascular disease risk in the Erqou meta-analysis, and the 58 nmol/L value that was reported for maximally reduced all-cause mortality risk in the Sawabe study:

Fortunately, I was able to reduce my Lp(a) levels from those first values to levels closer to ~100 nmol/L, which is still too high. For the first 8 Lp(a) measurements, I didn’t track my nutrition, so I can’t say which factors helped me to reduce it. Also, note that I didn’t include the blood test measurement where I tried high dose niacin (3 g/day), which reduced my Lp(a) to 84 nmol/L, but also worsened my liver function,. My liver enzymes, AST and ALT doubled on high-dose niacin! What good is a reduced risk for cardiovascular disease if my risk for liver disease simultaneously goes up? Obviously, I quickly discontinued use of niacin to reduce Lp(a).
Also note the data on the chart since 2015, when I started daily nutritional tracking. Over that period, my average value over 9 Lp(a) measurements is 95.3 nmol/L. Although my average Lp(a) is still higher than it should be, it’s better than my pre-tracking Lp(a) average value of 115.6 nmol/L (p-value = 0.03 for the between-group comparison). In addition, on my last 3 measurements, my Lp(a) values were 75, 82, and 79 nmol/L. How have I been reducing it?
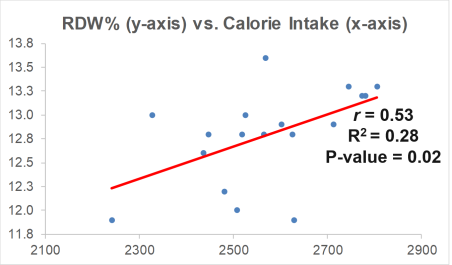
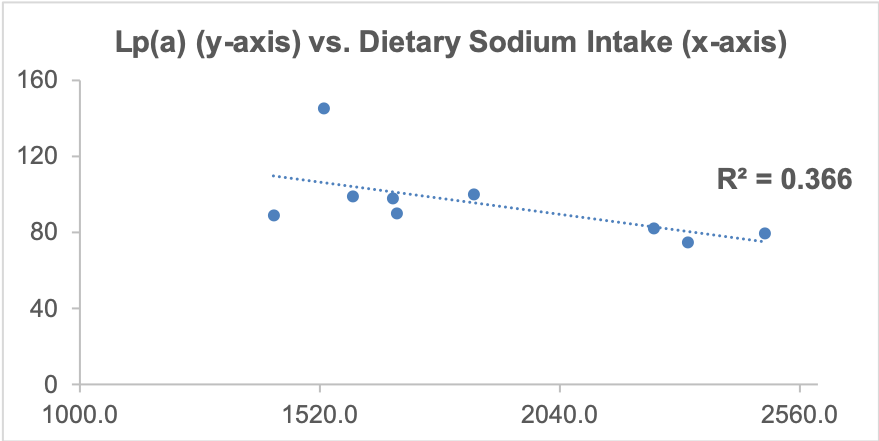
As I’ve mentioned in many blog posts, I’ve been weighing, logging, and tracking my nutrient intake since 2015. When I blood test, I can use the average dietary intake that corresponds to the blood test result, and with enough blood test results, I can look at correlations between my diet with blood test variables. Based on this approach, one possibility is my daily sodium intake. Shown below is a moderately strong correlation (r = 0.61, R^2 = 0.366) between my daily sodium intake with Lp(a). The higher my sodium intake, the lower my Lp(a) values.
Can the strength of this approach be improved? Interestingly, I identified another moderately strong correlation (r = 0.69) between my lycopene intake with Lp(a): the higher my lycopene intake, the higher my Lp(a)! I then decided to include both sodium and lycopene in a linear regression model, and the correlation for both of these nutrients with Lp(a) is 0.90! So what will I do with this info?
The highest that my average dietary sodium intake has been in any blood testing period is ~2500 mg. Sodium levels higher than that seem to negatively affect my sleep, so I’m not interested in going higher than 2500 mg/day. Also, there may be a plateau effect for sodium, as values ~2500 mg/day didn’t associate with significantly lower Lp(a) values when compared with 2300 mg/day. I can, in contrast, reduce my lycopene intake, which comes almost exclusively from my daily watermelon intake. I usually eat ~7 oz/day, and for my next blood test I’ll reduce this to 5 oz/day. Based on the regression equation that includes sodium and lycopene, with a 2300 mg sodium intake and the amount of lycopene that corresponds to 5 oz. of daily watermelon (~6700 micrograms, down from ~9000 micrograms), I should expect to see a Lp(a) value ~67 nmol/L on my next blood test. If not, I’ll repeat this approach, looking for strong correlations between my diet with Lp(a), followed by tweaking my diet to obtain biomarker results that are close to optimal. Stay tuned my my next blood test data, coming in about 2 weeks!
If you’re interested, please have a look at my book!
References
Clarke, R., J. F. Peden, J. C. Hopewell, T. Kyriakou, A. Goel, S. C. Heath, S. Parish, S. Barlera, M. G. Franzosi, S. Rust, et al. 2009. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N. Engl. J. Med. 361: 2518–2528.
Erqou, S., S. Kaptoge, P. L. Perry, A. E. Di, A. Thompson, I. R. White, S. M. Marcovina, R. Collins, S. G. Thompson, and J. Danesh. 2009. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 302: 412–423.
Langsted A, Kamstrup PR, Nordestgaard BG. High lipoprotein(a) and high risk of mortality. Eur Heart J. 2019 Jan 4. [Epub ahead of print].
Sawabe M, Tanaka N, Mieno MN, Ishikawa S, Kayaba K, Nakahara K, Matsushita S; JMS Cohort Study Group. Low Lipoprotein(a) Concentration Is Associated with Cancer and All-Cause Deaths: A Population-Based Cohort Study (The JMS Cohort Study). PLoS One. 2012; 7(4): e31954. PLoS One. 2012;7(4):e31954.
Nordestgaard BG, Chapman MJ, Ray K, Borén J, Andreotti F, Watts GF, Ginsberg H, Amarenco P, Catapano A, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, Lesnik P, Masana L, Reiner Z, Taskinen MR, Tokgözoglu L, Tybjærg-Hansen A; European Atherosclerosis Society Consensus Panel. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010 Dec;31(23):2844-53.